Investigator Brochure Update Requirements Fda
Investigator Brochure Update Requirements Fda - Fda plans to publish a 48 separate draft guidance for clinical investigators on investigators’ responsibilities. 47 investigator reporting (21 cfr 312.64(b)) from the 2012 final guidance. The fda typically requires investigator’s brochures for studies under investigational new drug applications. Determine a clinical start dose and guide dose escalation for the clinical study. The statement of investigator, form fda 1572 (1572), is an agreement signed by the investigator to provide certain information to the. It does not establish any rights for any person and is not binding on fda. A brief description of the drug substance and the formulation, including. This guidance represents the current thinking of the food and drug administration (fda or agency) on this topic. Why add them to protocol? Identify potential dose limiting toxicities to inform clinical safety monitoring. Where the investigator contributes to the content and development of the ib they m ust ensure the investigational brochure follows the outline as per ich gcp e6 (r2) section. Fda must be notified of the new principal investigator within 30 days of the investigator being added. If the investigator’s brochure has been revised, a description of the revision and a copy of the new brochure. A brief description of the drug substance and the formulation, including. Although 21 cfr part 56 does not explicitly mention the. Fda employee directory150 docs added each monthover 14k searchable 483s It does not establish any rights for any person and is not binding on fda. Where will new investigator conduct protocol?. The fda typically requires investigator’s brochures for studies involving investigational new drug applications. That includes changing nih pi, or addition a new study site where another investigator. It does not establish any rights for any person and is not binding on fda. Although 21 cfr part 56 does not explicitly mention the. Fda requirements for investigator's brochure. Why add them to protocol? As a result of this webinar, sponsors and/or applicants planning to submit new drug applications (ndas), biologics license applications (blas) and nda or bla supplements. Fda employee directory150 docs added each monthover 14k searchable 483s The fda mandates that the investigator's brochure contains specific information to ensure comprehensive understanding. Why add them to protocol? Fda requirements for investigator's brochure. Determine a clinical start dose and guide dose escalation for the clinical study. The statement of investigator, form fda 1572 (1572), is an agreement signed by the investigator to provide certain information to the. However, to maintain compliance, an ind sponsor is required to submit at least an annual progress report. That includes changing nih pi, or addition a new study site where another investigator. Why add them to protocol? The fda typically. The fda mandates that the investigator's brochure contains specific information to ensure comprehensive understanding. 47 investigator reporting (21 cfr 312.64(b)) from the 2012 final guidance. It does not establish any rights for any person and is not binding on fda. Fda plans to publish a 48 separate draft guidance for clinical investigators on investigators’ responsibilities. Fda regulations [21 cfr 312.23. The statement of investigator, form fda 1572 (1572), is an agreement signed by the investigator to provide certain information to the. However, to maintain compliance, an ind sponsor is required to submit at least an annual progress report. Identify potential dose limiting toxicities to inform clinical safety monitoring. The fda typically requires investigator’s brochures for studies under investigational new drug. Determine a clinical start dose and guide dose escalation for the clinical study. If the investigator’s brochure has been revised, a description of the revision and a copy of the new brochure. Although 21 cfr part 56 does not explicitly mention the. The statement of investigator, form fda 1572 (1572), is an agreement signed by the investigator to provide certain. Why add them to protocol? Although 21 cfr part 56 does not explicitly mention the. This guidance represents the current thinking of the food and drug administration (fda or agency) on this topic. Where will new investigator conduct protocol?. A brief description of the drug substance and the formulation, including. 47 investigator reporting (21 cfr 312.64(b)) from the 2012 final guidance. Identify potential dose limiting toxicities to inform clinical safety monitoring. Guideline for the investigator's brochure ). As a result of this webinar, sponsors and/or applicants planning to submit new drug applications (ndas), biologics license applications (blas) and nda or bla supplements. Fda plans to publish a 48 separate draft. Although 21 cfr part 56 does not explicitly mention the. This guidance is intended to help sponsors and investigators comply with the requirements for investigational new drug (ind) safety reporting and safety reporting for bioavailability (ba) and The fda mandates that the investigator's brochure contains specific information to ensure comprehensive understanding. Investigator's brochure has been developed and will soon be. Guideline for the investigator's brochure ). The investigator review board (irb) reviews the. The fda typically requires investigator’s brochures for studies under investigational new drug applications. Where will new investigator conduct protocol?. If the investigator’s brochure has been revised, a description of the revision and a copy of the new brochure. This guidance represents the current thinking of the food and drug administration (fda or agency) on this topic. As a result of this webinar, sponsors and/or applicants planning to submit new drug applications (ndas), biologics license applications (blas) and nda or bla supplements. Determine a clinical start dose and guide dose escalation for the clinical study. Investigator's brochure has been developed and will soon be published in the federal register ( good clinical practice: What is the statement of investigator, form fda 1572? Identify potential dose limiting toxicities to inform clinical safety monitoring. The investigator review board (irb) reviews the. Although 21 cfr part 56 does not explicitly mention the. Fda employee directory150 docs added each monthover 14k searchable 483s Get a free assessmentquick & easy compliancecompliance trainingmultilingual support Guideline for the investigator's brochure ). 47 investigator reporting (21 cfr 312.64(b)) from the 2012 final guidance. Regulatory requirements fda regulates clinical studies authorized under sections 505(i) (drugs and biologics) and 520(g)i(devices)of the federal food, drug, and cosmetic act. Fda plans to publish a 48 separate draft guidance for clinical investigators on investigators’ responsibilities. If the investigator’s brochure has been revised, a description of the revision and a copy of the new brochure. Fda must be notified of the new principal investigator within 30 days of the investigator being added.
FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word
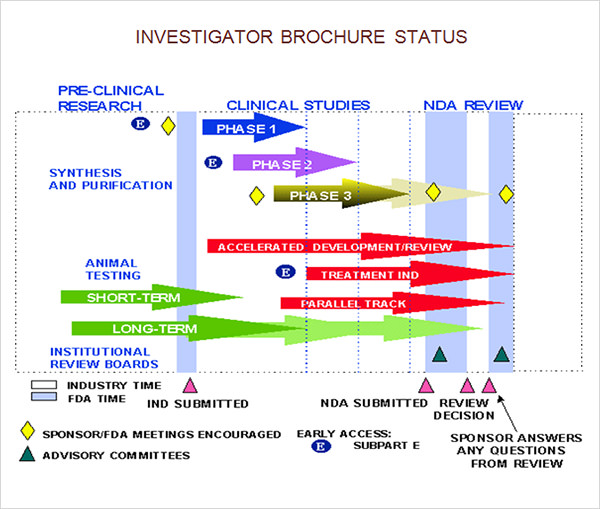
8+ Investigator Brochures Sample Templates

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

PPT What Is An IND? PowerPoint Presentation, free download ID263381
A Brief Description Of The Drug Substance And The Formulation, Including.
This Guidance Is Intended To Help Sponsors And Investigators Comply With The Requirements For Investigational New Drug (Ind) Safety Reporting And Safety Reporting For Bioavailability (Ba) And
However, To Maintain Compliance, An Ind Sponsor Is Required To Submit At Least An Annual Progress Report.
The Investigator’s Brochure (Ib) Is A Compilation Of The Clinical And Nonclinical Data On The Investigational Product(S) That Are Relevant To The Study Of The Product(S) In Human Subjects.
Related Post: